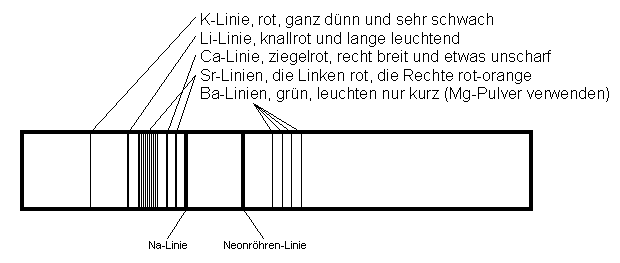
 |
| Kationen: | keine |
| Anionen: | Cl-, Br-, J-, CO32-, NO3-, PO43-, SO42- |
| Kationen: | Li+, Na+, K+, NH4+, Mg2+, Ca2+, Sr2+, Ba2+ |
| Anionen: | Cl-, NO3-, SO42- |
|
| Kationen: | Ag+, Hg1/2+, Pb2/4+, Bi3+, Cu+/2+, Cd2+, As3+/5+, Sb3+/5+, Sn2/4+ |
| Anionen: | Cl-, NO3-, SO42- |
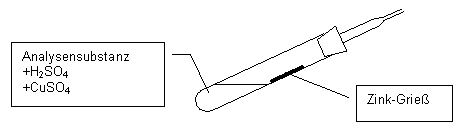 |
| Kationen: | Co2+, Ni2+, Fe2/3+, Mn2/4/7+, Al3+, Cr3/6+, Zn2+, Ti4+ |
| Anionen: | Cl-, NO3-, SO42- |
| Kationen: | Li+, Na+, K+, NH4+, Mg2+, Ca2+, Sr2+, Ba2+, Ag+, Hg1/2+, Pb2/4+, Bi3+, Cu+/2+, Cd2+, As3+/5+, Sb3+/5+, Sn2/4 +, Co2+, Ni2+, Fe2/3+, Mn2/4/7+, Al3+, Cr3/6+, Zn2+, Ti4+ |
| Anionen: | Cl-, NO3-, SO42-, PO43- |
| Kationen: | Li+, Na+, K+, NH4+, Mg2+, Ca2+, Sr2+, Ba2+, Ag+, Hg1/2+, Pb2/4+, Bi3+, Cu+/2+, Cd2+, As3+/5+, Sb3+/5+, Sn2/4 +, Co2+, Ni2+, Fe2/3+, Mn2/4/7+, Al3+, Cr3/6+, Zn2+, Ti4+ |
| Anionen: | Cl-, NO3-,
SO42-, PO43-
MoO42-, WO42-, VO43- |
| Kalium mit Perchlorsäure: | K+ + HClO4 --> KClO4 + H+ |
| Ammonium als NH3: | NaOH + NH4+ --> Na+ + NH3 + H2O |
| Quecksilber mit Münze: | Cu + Hg2+ --> Cu2+ + 2 Hg |
| Quecksilber als Iodid: | Hg2+ + 2 J- --> HgJ2 (rot) |
| Cobalt mit NH4SCN: | 4 NH4SCN + Co2+ --> H2[Co(SCN)4] (blau) + 4 NH4+ |
| Mangan mit PbO2: | 2 Mn2+ + 3 H2O + 5 PbO2 --> 2 MnO4- + 6 H+ + 5 PbO |
| Eisen mit NH4SCN: | Fe3+ + 3 SCN- --> Fe(SCN)3 (rot) |
| Cadmium als Sulfid: | Cd2+ + S2- --> CdS (gelb) |
| Zink als Sulfid: | Zn2+ + S2- --> ZnS (weiß) |
| Rinmanns Grün: | ZnO + 2 Co(NO3)2 --> ZnCo2O4 + 4 NO2 + 1/2 O2 |
| Thenards Blau: | Al2O3 + Co(NO3)2 --> 2 NO + 1/2 O2 + CoAl2O4 |
| Nitrat mit Devarda: | NO3- + 4 Zn + 7 OH- + 6 H2O --> NH3 + 4 [Zn(OH)4]2- |
| Chlorid mit AgNO3: | Ag+ + Cl- --> AgCl |
| Sulfat als BaSO4: | BaCl2 + SO42- --> BaSO4 + 2 Cl- |
| Phosphat: | 3 Zr4+ + 4 PO43- --> Zr3(PO4)4 |
| Soda-Pottasche für Ba: | BaSO4 + Na2CO3 --> BaCO3 + Na2SO4 |
| Soda-Pottasche für Alu: | Al2O3 + Na2CO3 --> 2 NaAlO2 + CO2 |
| Saurer Aufschluss: | Fe2O3 + 6 KHSO4 --> Fe2(SO4)3 + 3 K2SO4 + 3 H2O |
| Freiberger Aufschluss: | 2 SnO2+ 2 Na2CO3 + 9 S --> 2 Na2Sn3 + 3 SO2 + 2 CO2 |
| Oxidationsschmelze: | 2 FeCr2O4 + 4 K2CO3 + 7 NaNO3 --> Fe2O3 + 4 K2CrO4 + 7 NaNO2 + 4 CO2 |